Alternative splicing is a widespread post-transcriptional regulation mechanism that produces various mRNA transcript isoforms. Accurate identification of transcript types and their abundance is essential for studying the biological functions of alternative splicing. Emerging long-read sequencing technologies (LRS), capable of producing reads ranging from thousands to tens of thousands of bases, offer the potential to overcome the bottlenecks of precise transcript identification faced by short-read sequencing.
In recent years, numerous new bioinformatics tools have been developed to reconstruct transcript structures using LRS data. These algorithms can be categorized into two main types: guided and unguided. Guided algorithms require known reference gene annotations to assist in the transcript identification process, whereas unguided algorithms perform de novo reconstruction based solely on the sequence data, without needing reference annotations. Various strategies are employed by these tools to enhance detection accuracy, such as constructing splicing graphs, utilizing support read abundances, and applying machine learning models. Among them, TALON and FLAMES utilize a guided approach, whereas Freddie, TAMA, and UNAGI operate in an unguided mode. Meanwhile, StringTie2, FLAIR, IsoQuant, and Bambu include algorithms of both types. However, due to a lack of systematic evaluation, the performance of these tools under different data types and parameter settings remains unknown.
On May 10, 2024, Nature Communications published the latest research advancements from the team of researcher Dr. Wanlu Liu at the Zhejiang University-University of Edinburgh Institute (ZJE), titled "Comprehensive assessment of mRNA isoform detection methods for long-read sequencing data."

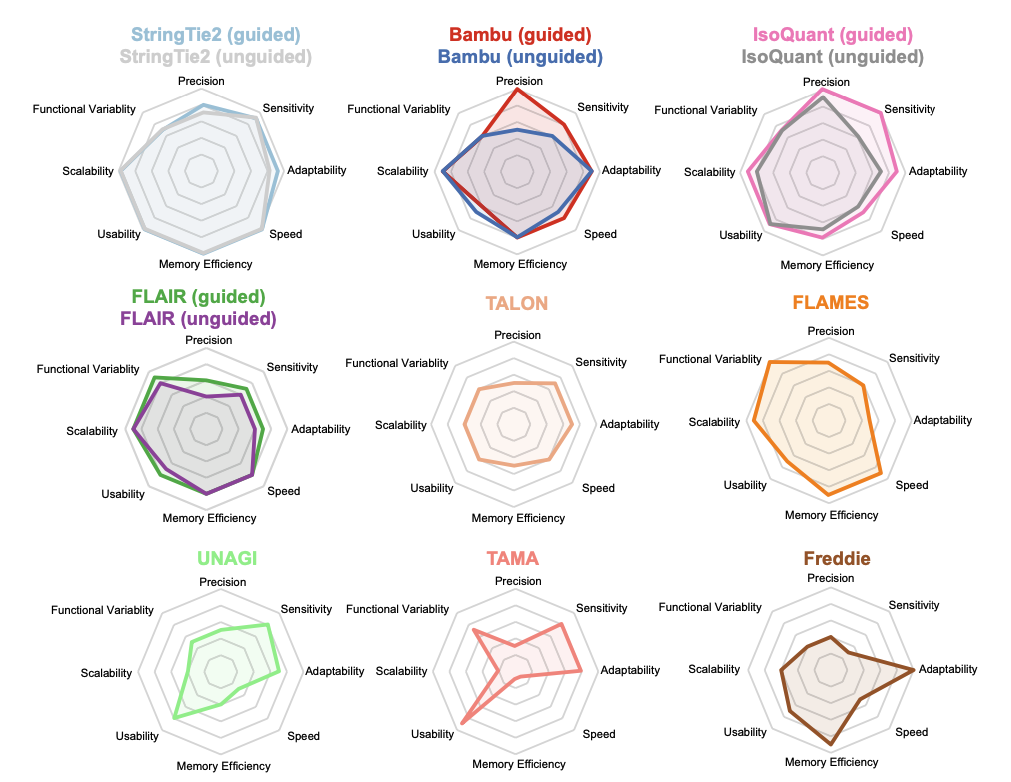
The study integrates various simulated and experimental data covering a wide range of biological scenarios, to conduct a comprehensive and in-depth evaluation of nine long-read mRNA isoform detection tools. The results demonstrate that IsoQuant is currently the most outstanding transcript identification tool, with Bambu and StringTie2 also showing impressive performances. Additionally, FLAMES and FLAIR are functionally comprehensive, supporting both upstream and downstream analyses. The authors analyzed the advantages and limitations of each algorithm and provided suggestions for improvement, directing future method development. This research establishes a foundation for fully exploiting long-read technologies to decipher transcriptome complexity. Concurrently, the authors developed the YASIM (Yet Another SIMulator) for long-read RNA sequencing, capable of simulating new alternative splicing events and real gene expression profiles and allowing precise control over various parameters such as sequencing depth, transcript complexity, read completeness, and sequencing accuracy to facilitate a comprehensive evaluation of the robustness of different tools.
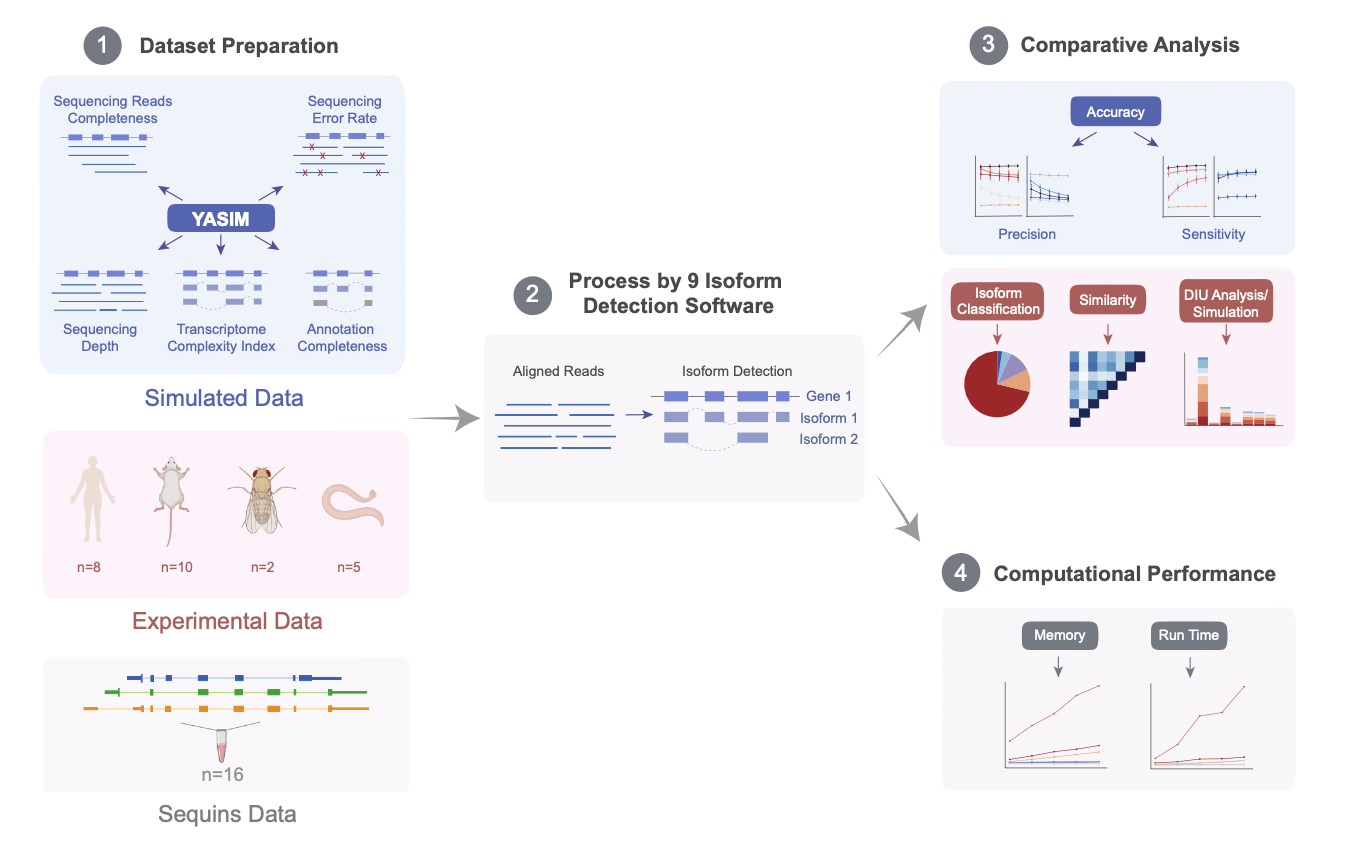
Initially, the authors used YASIM to generate simulated data under various conditions to assess the precision and sensitivity of different algorithms concerning sequencing depth, transcript complexity, read completeness, sequencing accuracy, and reference annotation completeness. The results indicated that IsoQuant and Bambu, under guided mode, showed the highest precision; IsoQuant and StringTie2 in guided mode demonstrated the best sensitivity. Most algorithms suffered degraded performance with shorter reads or lower sequencing accuracy.
Subsequently, the authors analyzed 16 published spike-in control datasets, and the results suggested that algorithms in guided mode outperformed those in unguided mode, with IsoQuant, Bambu, and StringTie2 in guided mode performing optimally overall. The authors also collected 25 real experimental datasets from four species for further detection analysis, which reconfirmed the high accuracy of IsoQuant and Bambu in guided mode for known transcript identification. Additionally, the authors conducted downstream analyses of differential isoform usage detected by various software, finding that the results from guided-mode tools were the most consistent, with IsoQuant and Bambu performing best. Based on mRNA isoform detection results from long-read sequencing data on a human embryonic stem cell line using StringTie2 and Bambu, the authors successfully validated a new isoform of the RPL39L gene through RT-qPCR experiments.
Finally, a computational efficiency analysis revealed that StringTie2 had the fastest run time and the lowest memory usage, with FLAMES, FLAIR, Bambu, and IsoQuant also performing well.
In summary, this study conducted a systematic evaluation of the main existing tools for mRNA isoform identification using LRS data by integrating simulation data, sequins control data, and experimental data. This evaluation work provides a reference for tool selection and highlights the strengths and weaknesses of various algorithms, facilitating the further optimization of long-read data mRNA isoform detection algorithms and promoting the application of long-read technologies in unraveling transcriptome complexity.
Yaqi Su, a 2019 undergraduate in Bioinformatics at ZJE (currently a PhD student at UC Berkeley), is the first author of this article, and Zhejian Yu, a 2019 undergraduate in Bioinformatics at ZJE (now a master’s student at the University of Edinburgh), is the second author. Dr. Wanlu Liu is the corresponding author. Other contributing authors include Siqian Jin, Ziwei Xue, Yixin Guo (doctoral students in bioinformatics at ZJE), Xinyi Chen (master's student in bioinformatics at ZJE), Zhipeng Ai (doctoral student in Zhejiang University School of Medicine), Ruihong Yuan (2019 undergraduate in Bioinformatics at ZJE), Dr. Di Chen (researcher at ZJE), Dr. Hongqing Liang (researcher at Zhejiang University School of Medicine), and Dr. Zuozhu Liu (researcher at the ZJUI institute).





