Decoding the tissue microenvironment and identifying potential therapeutic targets in cancer immunotherapy remain pressing frontiers of biomedical research. In recent years, spatial omics technologies have advanced rapidly. Spatial transcriptomics (ST) enables gene expression profiling within the spatial context of tissues, while spatial metabolomics (SM) uses mass spectrometry imaging to capture metabolite distributions. The integration of ST and SM offers multidimensional insights into the tissue microenvironment, particularly useful for uncovering metabolic heterogeneity and immune microenvironmental features in tumors. However, effective cross-modal and cross-sample integration has been hindered by fundamental differences in data type, spatial resolution, and tissue processing between ST and SM.
Recently, Dr. Wanlu Liu’s research group at the Zhejiang University-University of Edinburgh Institute (ZJE) published a study in Nature Communications titled: “Integrating cross-sample and cross-modal data for spatial transcriptomics and metabolomics with SpatialMETA.” The team developed a next-generation spatial multi-omics integration algorithm, SpatialMETA, which simultaneously achieves cross-modal and cross-sample integration of ST and SM data.
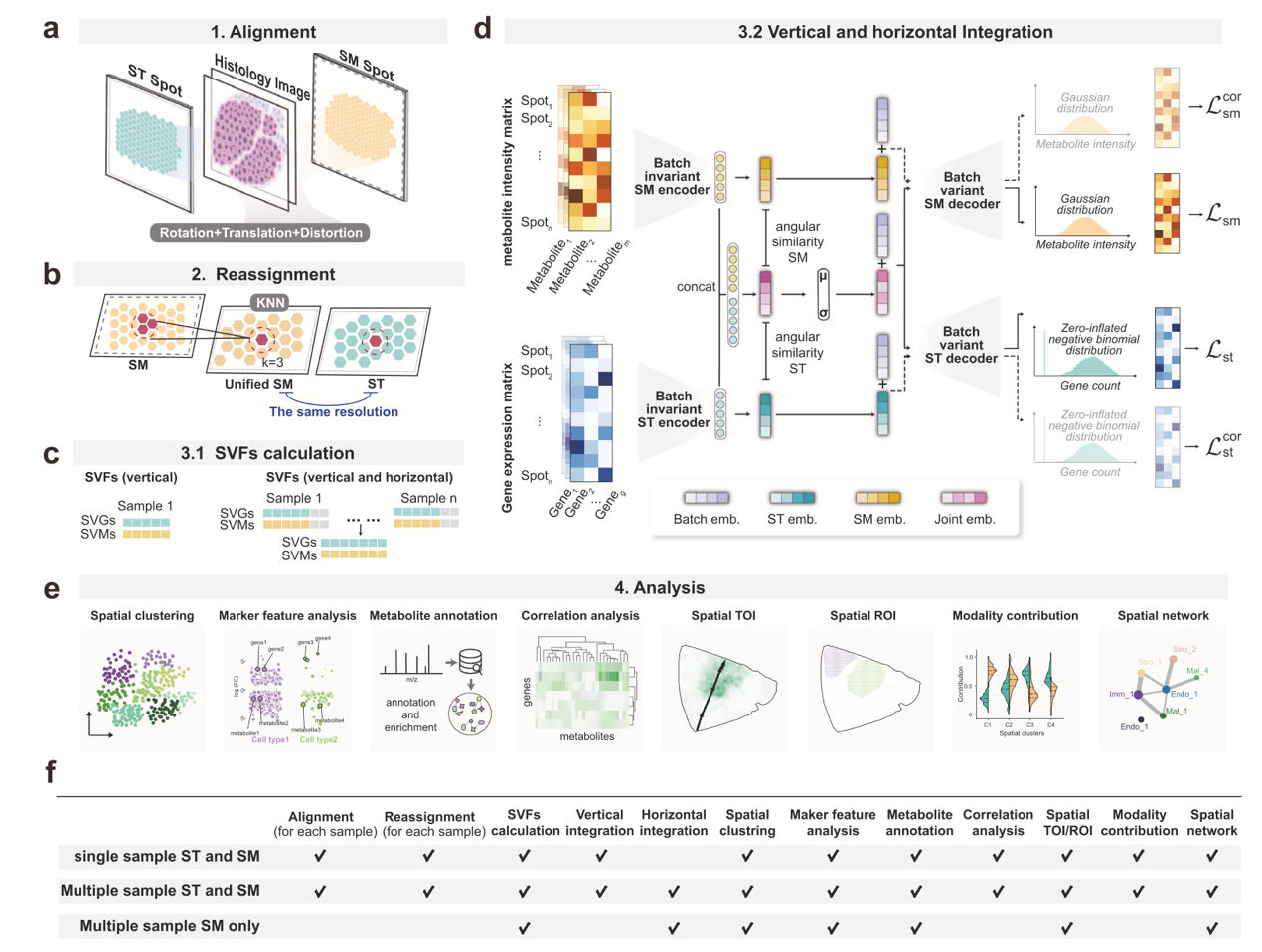
The method is built upon a conditional variational autoencoder (CVAE) framework, featuring tailored decoders and loss functions for ST and SM to accurately capture modality-specific feature distributions. SpatialMETA not only enables fusion across ST and SM modalities but also corrects batch effects between samples, facilitating consistent clustering across multiple datasets. Moreover, the model quantifies the contributions of different modalities to the integrated representation, thereby enhancing the biological interpretability of integration results.
The research team applied SpatialMETA to publicly available multi-omics datasets, including clear cell renal cell carcinoma (ccRCC), glioblastoma (GBM), and mouse brain. Results demonstrated that SpatialMETA accurately reconstructed the feature distributions of ST and SM and successfully identified immune-associated spatial clusters with unique metabolic features. These findings provide novel perspectives for understanding metabolic heterogeneity within tumor microenvironments. Compared with existing single-cell or spatial multi-omics integration tools, SpatialMETA outperforms in reconstruction accuracy, cross-modal fusion, and cross-sample consistency.
In addition, SpatialMETA offers a wide range of practical analysis modules, including automatic metabolite annotation based on m/z values, characterization of cluster-specific marker genes and metabolites, and correlation analysis between user-defined genes and metabolites. Notably, even with SM-only datasets, SpatialMETA supports cross-sample integration and analysis, expanding its applicability to diverse research scenarios.
Authors and Funding Information:
Dr. Wanlu Liu from the Zhejiang University-University of Edinburgh Institute (ZJE) is the corresponding author of this work. PhD candidates Ruonan Tian and ZJU-UoE dual-degree PhD candidates Ziwei Xue from ZJE are co-first authors. This research was conducted in collaboration with Professor Di Wang from Zhejiang University School of Medicine, Professor Jia Liu from the Shanghai Institute of Materia Medica, and Professor Youqiong Ye from Shanghai Jiaotong University. ZJE undergraduates Yiru Chen and Yicheng Qi also actively contributed to this study. This work has been supported by the National Key R&D Program of China, National Natural Science Foundation of China, the ZJU-YST joint research center for fundamental science, and the State Key Laboratory (SKL) of Biobased Transportation Fuel Technology.





